Substrate specificity and mismatch discrimination in DNA ligases
by Greg Lohman, Ph.D., New England Biolabs, Inc.
DNA ligases vary in their ability to join fragments, add adaptors, repair nicks or breaks, link vectors and inserts, and to circularize dsDNA. Ligases also vary in their type of activity. The specificity and accuracy of the ligation depends upon ligase selection and careful optimization of reaction conditions. With the right ligase, conditions and probes, even single-base variations in sequence can be reliably detected.
DNA ligases are enzymes that seal breaks in DNA by joining 5´-phosphorylated DNA termini to 3´-OH DNA termini (1-4). In vivo, ligases are important for the repair of nicks, breaks in one strand of a dsDNA molecule, DNA formed during replication (i.e., Okazaki fragments), as well as both nick and double-strand break joining during repair events (5). In vitro, ligases (notably T4 DNA Ligase) are critical reagents for many molecular biology protocols, including vector-insert joining for recombinant plasmid construction, adaptor ligation for next-generation sequencing (NGS) library construction, and circularization of dsDNA (6). T4 DNA Ligase (NEB #M0202) is incredibly efficient at sealing nicks, as well as joining or circularizing DNA fragments with blunt or cohesive (short complementary) ends. This activity can be further improved with the addition of macromolecular enhancers, such as polyethylene glycol (PEG), as seen in NEB’s Quick Ligation™ Kit (NEB #M2200) (7,8).
(Watch the video.)
Less commonly utilized in vitro, Taq DNA Ligase (NEB #M0208) will ligate only nicks (9-12). Taq Ligase is a NAD+-dependent DNA ligase from a thermostable bacterium that can survive high temperatures (up to 95°C) and is active over a range of elevated temperatures (37–75°C). However, it only has significant activity on nicked DNA, and negligible activity on short cohesive and blunt substrates in end-joining reactions. Given these limitations, and the fact that T4 DNA Ligase can ligate everything Taq ligates, and many more structures, why not use T4 DNA Ligase for all applications?
T4 DNA Ligase can ligate a wide variety of DNA structures, including modified bases and the ends of double stranded fragments. It will also efficiently ligate many undesirable structures, including substrates containing gaps of one or more nucleotides and nicked substrates that contain DNA base pair mismatches (12-15). In most cases, this unwanted activity isn’t a problem, for example, when joining 1 or 2 fragments into a plasmid, or pushing an adaptor ligation reaction as far towards completion as possible to prepare high yields of DNA NGS libraries.
For some applications, however, there cannot be any end-joining activity at all, and for others, there is a need for the exclusive ligation of fully base-paired nicks with no gaps. For example, DNA assembly methods, such as Gibson Assembly® (NEB #E5510) and NEBuilder® HiFi DNA Assembly (NEB #E2621), require nick-selective ligases; this method utilizes long overlaps that are dynamically generated by exonucleases and gaps are filled by a DNA polymerase (16,17). Final joining is accomplished by a nick-selective ligase, such as Taq DNA Ligase, which only reacts with substrates containing no gaps, and will not join any fragments end-to-end without the exo/polymerase generation of annealed complemen-tary regions. The use of a nick-selective ligase ensures that fragments are not joined out of order, and no deletions result from ligation across nucleotide gaps in annealed structures. (For more information, see www.neb.com/DNAassembly).
Ligase Specificity
DNA ligases generally prefer fully Watson-Crick base-paired dsDNA substrates to those containing one or more mismatches. However, ligases can ligate some mismatches to a significant degree, and very active ligases, such as T4 DNA Ligase, can ligate nicks containing one or more mismatches near the ligation junction with high efficiency (15,18). Ligases are thought to interrogate dsDNA for proper base pairing through minor grove contacts, and thus do not read specific base sequences, but are sensitive to distortions in helix shape (19). Large purine:purine mismatches and most smaller pyrimidine:pyrimidine mismatches are typically worse ligation substrates than pyrimidine:purine mismatches. Helix stability also plays some role, and mismatches with more hydrogen bonds are more readily ligated than those with few. For many ligases, G:T mismatches, with two hydrogen bonds and a base-pair size nearly indistinguishable from a Watson-Crick base pair, are joined with nearly the same efficiency as a correct base pair. Additionally, DNA ligases have been generally found to have a higher discrimination at the upstream side of the ligation junction (the base pair providing the 3´-OH terminus to the ligation) than on the downstream side (the base pair providing the 5´-phosphate to the ligation). The structural/mechanistic reason for this differential is not known for certain, but may have to do with the slight melting of the 5´-terminus during the reaction. This “peeling back” of the 5´-phosphorylated base can be observed in the crystal structures of several DNA ligases bound to substrate (20,21).Thermostable DNA ligases, including Taq DNA ligase, are naturally able to discriminate against ligating substrates containing base pair mismatch-es (i.e., are "higher fidelity") than T4 DNA Ligase (18,22,23). Despite this higher fidelity, Taq DNA Ligase can still detectably ligate many T:G, T:T, and A:C mismatches. Thermostable DNA ligases are active at elevated temperatures, allowing fur-ther discrimination by incubating the ligation at a temperature near the melting temperature (Tm) of the DNA strands. This selectively reduces the concentration of annealed mismatched substrates (expected to have a slightly lower Tm around the mismatch) over annealed fully base-paired substrates. Thus, high-fidelity ligation can be achieved through a combination of the intrinsic selectivity of the ligase active site and careful balance of reaction conditions to reduce the incidence of annealed mismatched dsDNA.
Applications Requiring High-Fidelity Ligation
Numerous applications have been developed that take advantage of the high fidelity of Taq and other thermostable DNA ligases to detect specific nucleotide sequences with high specificity and quantitative accuracy, including profiling single nucleotide polymorphisms (SNPs) (9,24,25). In the Ligase Detection Reaction (LDR), a set of probes complementary to the sequence of interest are annealed to target DNA (genomic DNA, or a PCR amplified fragment) and treated with a high-fidelity thermostable DNA ligase (Figure 1). If the target sequence is present, the probes will ligate; cycling through rounds of melting and annealing can allow linear amplification of the probe ligation product. With the right ligase, conditions and suitable probes, single-base differences can be reliably detected. The original paper detected the ligation product through visualization in a gel, but detection through fluo-rophore-quencher pairs or qPCR-based methods can greatly increase the sensitivity of detection (26-32). LDR has also been extended to mul-tiplexed probe sets that allow the simultaneous interrogation of multiple potential SNP sites (27).
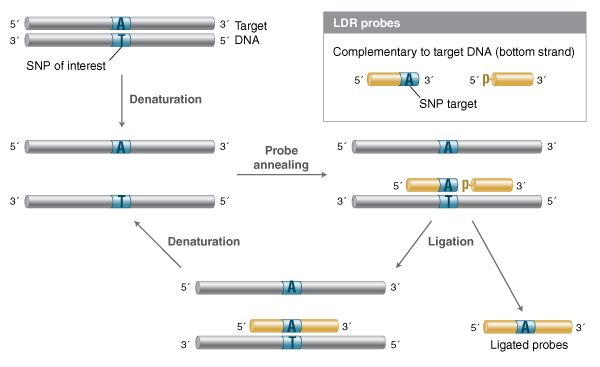
Two ligation probes are designed such that they are complementary to a target region of interest, and anneal with no gaps. Typically, if a SNP is to be resolved, the nucleotide of interest is situated at the junction of the two probes. The probes are combined with the DNA to be examined (typically genomic or PCR amplified region) and the thermostable DNA ligase. The DNA is melted, then cooled to a ligation temperature that al-lows the probes to anneal to the target. If the probes anneal to form a nicked structure with no gaps or mismatches, efficient ligation will proceed. Cycling, melting and annealing/ligation allows successive rounds of probes to anneal and ligate, resulting in linear amplification of the ligation product if the sequence of interest is present in the target DNA.
The closely related Ligase Chain Reaction (LCR) takes the LDR method and makes it amplifiable in an exponential fashion (9,24). In LCR, four probes are used, one pair complementary to one target strand, and a second pair of probes complementary to the other strand (Figure 2). Since the probe pairs are complementary to each other, the probe ligated in one cycle becomes a template for ligation of additional probe in subsequent cycles. This methodology allows for detection of SNPs with greater sensitivity than the original LDR method, but requires extraordinary discrimination against mismatch ligation for both probe sets, as even trace ligation on a mismatched template will result in template for further probe amplification (and thus, an erroneous positive signal). The complementarity of LCR probe pairs also means that probes can anneal to each other, forming blunt-end or single-base overhang substrates, depending on probe design strategy. While most high-fidelity ligases have far lower activity on double-stranded fragment end joining, even trace blunt-end activity will generate template for further rounds of high-efficiency nick ligation. Thus, LCR can suffer from high non-templated background as well, and requires careful probe design. The modification gap-LCR method attempts to address these background and discrimination issues by utilizing probes which anneal with a single-nucleotide gap that must be filled by a polymerase in order to generate a substrate suitable for ligation (33). This modification leverages the discrimination against gap ligation of thermostable high-fidelity ligases, but requires a thermostable polymerase and dNTPs as well.
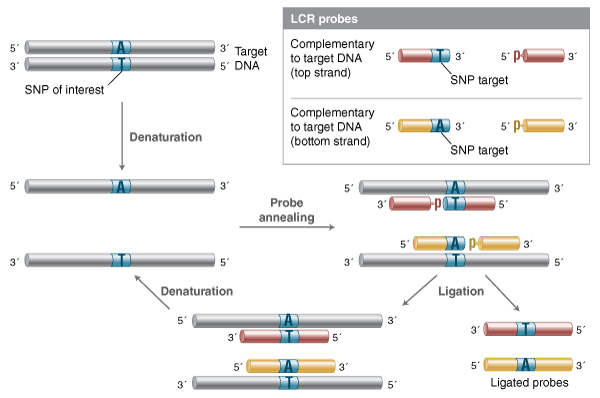
In this method, two pairs of probes are designed, one pair complementary to the top strand of the target, one pair to the bottom. Upon melting and annealing, both probe pairs can anneal to the target, and ligate efficiently if they form a nicked sequence without gaps or mismatches. On successive rounds of melting and re-annealing, unligated probe can now anneal to both the original target DNA and to the probes ligated in previous rounds. As each ligation product becomes a template for the complementary probe pair, LCR enables exponential amplification
of the ligated product.
Additional detection-by-ligation technologies have been devised to take advantage of high-fi-delity ligation events by generating circular templates that can be detected in a secondary reaction (34-38). In the “padlock” probe design, a single-stranded probe is devised where the 5´ and 3´ ends are both complementary to a target sequence (Figure 3).
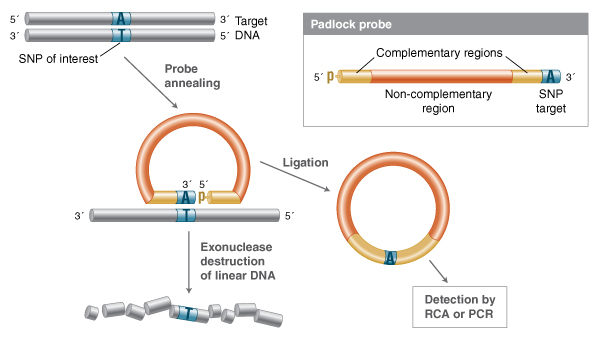
In this method, a single probe is designed such that the ends of the probes are complementary to the target sequence. When annealed to the de-sired target, the ends form a nicked structure that can be efficiently ligated if there are no gaps or mismatched base pairs. Exonuclease treatment destroys the uncircularized DNA, and the remaining circular structures can be detected through rolling circle amplification (RCA), or linearized and amplified with PCR.
Much like LDR, the ends of this single probe form a nick structure with no gaps when annealed, and can only be ligated when fully base-paired to a complementary target. All single-stranded DNA can be destroyed by exonuclease treatment, and the circularized probes can be detected by methods such as rolling circle amplification, or linearized and detected by PCR. Variation on the padlock probe design include “molecular inversion probes,” which, similar to gap-LCR, have a single-stranded probe in which both ends are annealed with a gap of one or more nucleotides that must be filled by a polymerase before ligation can occur.
Other Factors to Consider
These and other detection-by-ligation methodologies depend on the ability of the ligase to discriminate against substrates containing one or more mismatches, yet retain high activity on even low concentrations of the fully base-paired probe-target structure. While the choice of ligase is very important, careful probe design, selection of reaction temperature, and even ligation buffer conditions can all contribute to the fidelity of the ligation reaction, and thus the accuracy and sensitivity of the detection-by-ligation. For example, probes should take advantage of the naturally higher discrimination of ligases on the upstream side of the ligation junction (the base pair providing the 3´-hydroxyl). Probes that place the base of interest on the downstream side will provide significantly poorer discrimination, and probes will ligate on templates containing other bases at the targeted position. Furthermore, it is important to know what base pair mismatches are more easily ligated by a given ligase. For example, if you are targeting a position that can be an A:T or a T:A base pair, it would be better to use a probe with an A at the 3´ end (targeting the strand with a T at the SNP position) than to use a probe with a T; when annealing to the wrong SNP, the first case would result in a difficult-to-ligate A:A mismatch, while the second would result in a T:T mismatch that can be ligated by Taq DNA Ligase with relatively high efficiency (Figure 4).
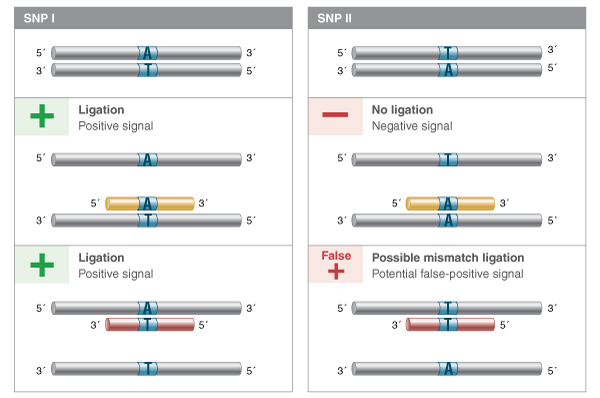
LDR or padlock probes can be designed to target either strand of a sequence of interest. If the SNP to be interrogated is a T:A or an A:T base pair, and a positive signal is desired from SNP I but not SNP II, it is better to use a probe set that pairs an A with the T in the target, as annealing to the alternate SNP will result in a difficult to ligate A:A base pair. If probes are designed such that a T in the probe pairs with an A in the sequence of interest, then the alternate SNP will form a T:T mismatch, more easily ligated by many thermostable ligases, and result in a false positive signal.
Incubation temperature is also a key consider-ation, and typically must be optimized for each application. If the ligation temperature is too far below the Tm of the probes, even mismatched probes will be annealed, increasing the chances of ligation occurring. If the ligation temperature is too far above the Tm, fully complementary sequences will not be annealed. High-fidelity ligation reactions should typically be run 1–2°C below the Tm of the probes to give the highest possible accuracy by minimizing the concen-tration of annealed mismatched probes. Conse-quently, it is important to match the Tm of both the upstream and downstream probe annealing regions, and all probe sets when attempting a multiplexed reaction. If there is a range of Tm values for the probe annealing regions, no single reaction temperature will result in the optimal balance of fidelity versus activity for all probe sets.
Buffer conditions can also affect the fidelity of DNA ligases. In particular, it has been observed for several thermostable DNA ligases, including T4 DNA Ligase and Human DNA Ligase 3, that increasing monovalent cation concentration improves the fidelity of ligation (15,39) . This effect is thought to be related to a weakening of the binding of the ligase to its substrate, with a disproportionate suppression binding/ligation of mismatched substrates. Too much salt can erase activity on even fully base-paired substrates, and the best salt balance for each ligase must be empirically determined.
Optimization Through High-Throughput Profiling
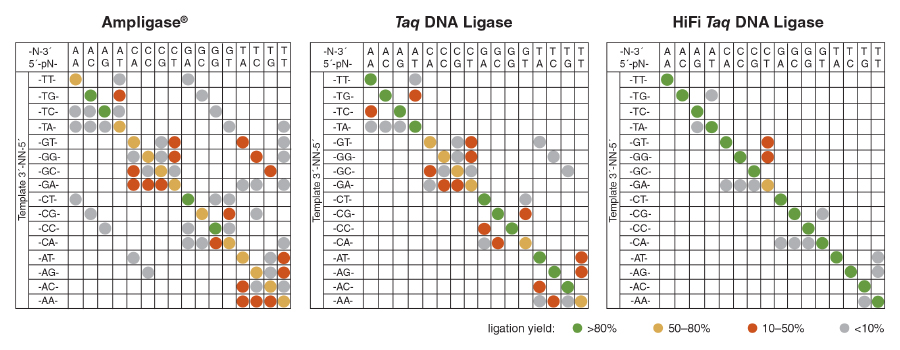
NEB researchers recently published a method for the high-throughput profiling of ligase fidelity, a method that extends earlier studies through a high-sensitivity multiplexed format (18,40). This methodology has been used to rapidly screen buffer conditions for Taq DNA Ligase and their effect on fidelity. In this method, substrate pools were prepared consisting of one target (template) strand and four upstream probes and four down-stream probes, each differing only in the base at the ligation junction. Thus, all four bases at either side of the ligation junction were represented. Sixteen separate pools were prepared, each with a different template strand covering all 16 possible NN pairs in the template as well. The probes were designed such that each possible pairing resulted in a product of unique length, with products repeatable and quantifiable by capillary electrophoresis (CE) (Figure 5). This method allowed screening of all possible base combina-tions (Watson-Crick and mismatched) around the ligation junction in 16 wells of a 96-well plate, allowing 6 conditions to be screened per plate. The results indicated that the optimal buffer for Taq DNA Ligase contains with 100 – 200 mM KCl at pH 8.5.

Each substrate pool contained a single splint with a defined NN at the ligation junction (e.g., AA, AC, AG…) along with all four upstream probes and all four FAM-labeled downstream probes. Each probe is of unique lengths that encode the base at the ligation junction: 20, 28, 36 and 44 bases for the 3´-A, C, G and T´ terminated upstream probes; 30, 32, 34 and 36 bases for the 5´-pA, pC, pG and pT terminated, 3´-FAM-la-beled downstream probes. A total of 16 substrate pools were prepared, one for each unique splint. Figure and figure caption reproduced from (Lohman, G.J. et al. 2016) under the creative commons license.
Conclusion
This optimization method has been used internally at NEB to screen additional ligases, conditions and formulations, and has led to the development of the new HiFi Taq DNA Ligase (NEB #M0647). Using this method, both the enzyme and the reaction buffer were optimized, resulting in the highest fidelity NAD+-dependent DNA ligase commercially available (Figure 6).
It is important to note that thermostable, high-fidelity, nick-selective DNA ligases like Taq DNA Ligase, HiFi Taq DNA Ligase, and the ATP dependent 9°N™ DNA Ligase (NEB #M0238), are not replacements for T4 DNA Ligase in applications such as routine cloning or DNA library preparation. However, when a method relies on accurate ligation of nicks lacking gaps or mismatched base pairs, using of one of these ligases, combined with careful probe design and reaction condition optimization, will be critical for success.
References
- Shuman, S. and C. D. Lima (2004). Curr. Opin. Struct. Biol. 14, 757-764.
- Tomkinson, A. E., et al. (2006). Chem. Rev. 106, 687-699.
- Ellenberger, T. and A. E. Tomkinson (2008). Annu. Rev. Biochem. 77, 313-338.
- Shuman, S. (2009). J. Biol. Chem. 284, 17365-17369.
- Arakawa, H. and G. Iliakis (2015). Genes (Basel) 6, 385-398.
- Lohman, G. J., et al. (2011). Curr. Protoc. Mol. Biol. Chapter 3: Unit 3.14.
- Pheiffer, B. H. and S. B. Zimmerman (1983). Nucleic Acids Res. 11, 7853-7871.
- Hayashi, K., et al. (1986). Nucleic Acids Res. , 7617-7631.
- Barany, F. (1991). Proc. Natl. Acad. Sci. USA 88, 189-193.
- Barany, F. and D. H. Gelfand (1991). Gene 109, 1-11.
- Lauer, G., et al. (1991). J. Bacteriol. 173, 5047-5053.
- Luo, J. and F. Barany (1996). Nucleic Acids Res. 24, 3079-3085.
- Nilsson, S. V. and G. Magnusson (1982). Nucleic Acids Res. 10, 1425-1437.
- Goffin, C., et al. (1987). Nucleic Acids Res. 15, 8755-8771.
- Wu, D. Y. and R. B. Wallace (1989). Gene 76, 245-254.
- Gibson, D. G., et al. (2009). Nat Methods 6, 343-345.
- Gibson, D. G., et al. (2010). Science 329, 52-56.
- Lohman, G. J., et al. (2016). Nucleic Acids Res. 44, e14.
- Liu, P., et al. (2004). Nucleic Acids Res. 32, 4503-4511.
- Pascal, J. M., et al. (2004). Nature 432, 473-478.
- Nandakumar, J., et al. (2007). Mol. Cell 26, 257-271.
- Luo, J., et al. (1996). Nucleic Acids Res. 24, 3071-3078.
- Nishida, H., et al. (2005). Acta. Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 1100-1102.
- Barany, F. (1991). PCR Methods Appl. 1, 5-16.
- Wiedmann, M., et al. (1994). PCR Methods and Applications 3, S51-64.
- Zirvi, M., et al. (1999). Nucleic Acids Res. 27, e40.
- Pingle, M. R., et al. (2007). J. Clin. Microbiol. 45, 1927-1935.
- Cheng, C., et al. (2013). Anal. Biochem. 434, 34-38.
- Hamada, M., et al. (2013). Electrophoresis 34, 1415-1422.
- Hommatsu, M., et al. (2013). Anal. Sci. 29, 689-695.
- LeClair, N. P., et al. (2013). J. Clin. Microbiol. 51, 2564-2570.
- Watanabe, S., et al. (2014). Anal. Chem. 86, 900-906.
- Marshall, R. L., et al. (1994). PCR Methods and Applications 4, 80-84.
- Nilsson, M., et al. (1994). Science 265, 2085-2088.
- Cao, W. (2001). Clin. Appl. Immun. Rev. 2, 33-43.
- Qi, X., et al. (2001). Nucleic Acids Res. 29, E116.
- Cao, W. (2004). Trends in Biotechnology 22, 38-44.
- Cheng, Y., et al. (2013). Analyst 138, 2958-2963.
- Bhagwat, A. S., et al. (1999). Nucleic Acids Res. 27, 4028-4033.
- Greenough, L., et al. (2016). Nucleic Acids Res. 44, e15.

